


![]()

![]()












Suscripción a nuestro boletín.








¿Qué es?
En este tipo de enfermedad las personas no pueden descomponer las proteínas correctamente. Esto causa la acumulación de sustancias dañinas en la sangre y en la orina. Éstas pueden afectar a la salud, el crecimiento y el aprendizaje. Se debe a la falta o la disfunción de la enzima "glutaril-CoA deshidrogenasa". El trabajo de esta enzima es procesar una sustancia llamada glutaril-CoA que se produce cuando se procesan los aminoácidos lisina, hidroxilisina y triptófano. Este defecto metabólico causa un acumulo de glutaril CoA. Ésta se esterifica en parte con carnitina, produciendo un déficit secundario de ésta, causa importante de las crisis metabólicas. La lisina y el triptófano se hallan en todas las comidas que contienen proteínas.
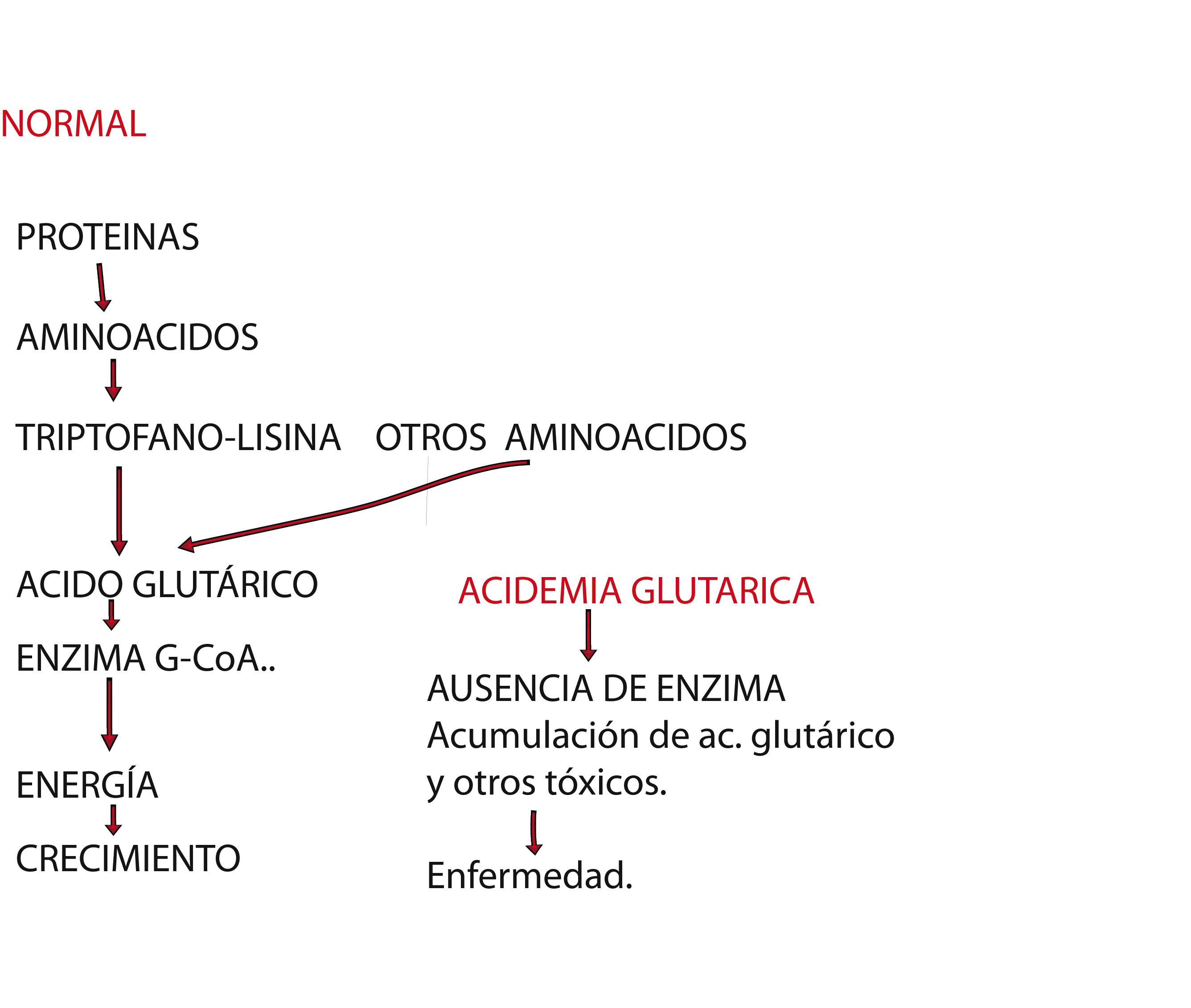
Es una enfermedad hereditaria.
¿Cómo se hereda?
Es autosómica recesiva. Los padres de niños con la enfermedad no siempre la padecen.
Cuando ambos padres son portadores, hay un 25% de probabilidad en cada embarazo de que el niño tenga la enfermedad. Hay un 50% de probabilidad de que el niño sea un portador, igual que los padres. Finalmente, hay un 25% de probabilidad de que el niño tenga dos genes que funcionan correctamente y esté sano.

¿Es una enfermedad frecuente?
Es rara.
La incidencia, es decir, el número de casos nuevos, es menor de 1:75.000. La mayor prevalencia se da en la comunidad Amish, los indios Oji-Cree de Canadá y en personas de ascendencia sueca.
¿Y los síntomas clínicos?
Los síntomas suelen comenzar entre los dos meses y los cuatro años de edad. El 74% de los casos que tienen más riesgo de crisis encefalopáticas, tienen desde que nacen una cabeza de mayor tamaño que la media. Estas crisis consisten en la aparición de:
convulsiones
disminución del nivel de conciencia
irritabilidad
hipotonía
dificultades para la alimentación
y la presencia de movimientos involuntarios denominados distónicos o coreicos. Se trata de contracciones musculares sostenidas que causan torceduras y movimientos repetitivos involuntarios que se traducen en posturas anormales. Pueden afectar a un solo músculo, a un grupo de músculos como los de los brazos, las piernas o el cuello, o incluso al cuerpo entero.
También puede causar episodios de enfermedad graves. Se llaman crisis metabólicas. Algunos de los síntomas son:
falta de apetito
demasiado sueño
falta de energía
irritabilidad
ansiedad
náuseas
vómitos
tono muscular bajo
debilidad muscular.
Si el niño no recibe tratamiento, pueden aparecer otros síntomas como:
tics o espasmos musculares
contracciones musculares rígidas (espasticidad)
movimientos violentos involuntarios de los brazos y las piernas
poca coordinación. Problemas de equilibrio
aumento de los niveles de sustancias ácidas en la sangre
convulsiones
inflamación o sangre en el cerebro
coma, que puede terminar en la muerte.
Las crisis metabólicas generalmente son causados por una descompensación que puede ser provocada por: una enfermedad o infección, la fiebre o el ayuno.
Otros efectos de la GA-1 que pueden suceder, incluso, sin una crisis metabólica son:
escaso crecimiento
aumento del tamaño del hígado
flacidez muscular
rigidez muscular progresiva
movimientos involuntarios musculares
episodios repetidos de fiebre
sudoración excesiva
retrasos para caminar y de otras habilidades motoras
retrasos en el aprendizaje y retraso mental
problemas del lenguaje
daño cerebral.
Algunas personas tienen síntomas muy leves o no tienen síntomas. Y solo se descubre que tienen la enfermedad después de haberla diagnosticado a un hermano o hermana.
¿Y el cribado neonatal?
Es muy importante para el diagnóstico precoz.
Se hace cribado universal, es decir el estudio de toda la población. Se realiza con la determinación de glutarilcarnitina (C5-DC) y el ratio C5-DC/C16 en sangre impregnada en papel recogida del talón del recién nacido.
Cuando ambos, el padre y la madre son portadores, los resultados de las pruebas de detección sistemática en el recién nacido no son suficientes para descartar la GA-1.
En estos casos, se deben hacer pruebas de diagnóstico especiales además de las pruebas de detección sistemáticas propias del recién nacido.
¿Y el diagnóstico de confirmación?
Se realiza con:
Cuantificación de los ácidos orgánicos en orina de una micción aislada.
Cuantificación de acilcárnitinas en plasma y orina
Estudio de la actividad enzimatica de la glutaril-CoA deshidrogenasa en fibroblastos de piel
Análisis molecular del gen GCDH. En sangre anticoagulada
¿Es posible el diagnóstico prenatal?
Si.
Si hay alteraciones en ambos genes del paciente, se pueden analizar el ADN en embarazos futuros.
Los padres pueden elegir hacer los estudios de detección durante el embarazo o esperar hasta el nacimiento.
¿Y el tratamiento?
Los niños con síntomas de crisis metabólica necesitan tratamiento médico de inmediato. Generalmente en el hospital. Durante una crisis metabólica, los niños pueden recibir líquidos, glucosa, insulina, carnitina y otros medicamentos por vía intravenosa, para ayudarlos a eliminar las sustancias dañinas de la sangre.
Evite que el niño pase demasiado tiempo sin comer. Por lo general, se suele sugerir alimentar a los niños cada cuatro a seis horas. Algunos bebés necesitan comer con mayor frecuencia inclusive. Es importante que los bebés coman durante la noche.
Cuando están bien, muchos niños de mayor edad y adultos con GA-1 pueden pasar hasta 12 horas sin comer. Pero pueden necesitar seguir con los otros tratamientos de por vida.
El tratamiento dietoterapéutico se hace con base en la restricción de proteínas de alto valor biológico natural. Es decir aquellas que están en la carne, pescado, etc. Y suplementación de carnitina.
La mayor parte de los niños necesita una dieta constituida por alimentos bajos en lisina y triptófano. Generalmente los alimentos médicos especiales y una fórmula especial son parte de la dieta.
Es muy importante que el núcleo familiar mantenga una adecuada adherencia al tratamiento. De esta forma se logra un buen control metabólico que permite evitar la descompensación y las crisis subsiguientes.
La educación nutricional se enfoca en establecer buenos hábitos desde un inicio. Variar la dieta ofreciendo todo tipo de frutas y mezclas de ellas, para lograr introducir la mayor cantidad de sabores y texturas. La alimentación futura de estos niños se basa, fundamentalmente, en frutas y verduras.
¿Es posible que otros miembros de la familia padezcan la enfermedad?
Los hermanos o hermanas tienen probabilidades de tener la enfermedad, aunque no hayan tenido síntomas.
Pueden ser portadores como sus padres.
Los hermanos de cada padre tienen un 50% de probabilidad de ser portadores.
Enlaces útiles

![]()
![]()
 Compartir
Compartir






